In diesem Beitrag wollen wir uns näher mit den Konsequenzen mitochondrialer Krankheiten beschäftigen. Bei der Recherche zum Thema konnte ich feststellen, dass es in der Tat viele Ansichten gibt, wann man von einer mitochondrialen Dysfunktion überhaupt sprechen kann. Die einen sehen eine mitochondriale Dysfunktion auf Grundlage von Mutationen in mit Mitochondrien-assoziierten Genen an, welche in ihrer Endkonsequenz die Funktionsfähigkeit der Mitochondrien mindern. Andere fassen die Problematik etwas weiter (so auch ich) und würden eher alle Beeinträchtigungen der Zelle, welche letztlich die Leistung der Mitochondrien herabsetzen miteinbeziehen.
Mitochondriale Dysfunktion sind, sofern sie vererbt wurden, mit erheblichen Problemen für den Menschen verbunden [1, 2]. Das bedeutet, dass die meisten Menschen entweder überhaupt nicht lebensfähig sind (also früh in der Entwicklung oder spätestens im Kindesalter versterben) oder spätestens als Erwachsene erhebliche gesundheitliche Probleme haben. Beispiele für solche Probleme sind unter anderem Taubheit, Blindheit, Herzprobleme, Diabetes, Leberschäden, Muskelschwächen und allgemeine Erschöpfung bis hin zu neurologischen Problemen wie Depressionen und Demenz [1-6].
Da viele der Erkrankungen multifaktorielle sind, ist es allerdings sehr schwierig einzuschätzen woher die Probleme hauptsächlich stammen. Die Kenntnis über die Ursache einer Erkrankung ist aber absolut entscheidend, um eine konkrete therapeutische Konsequenz abzuleiten zu können. Und Mitochondrien sind prädestiniert für solche multifaktoriellen Probleme, denn wie ihr aus dem ersten Teil wisst, sind diese eifrigen Gefährten an eigentlich allem was in der Zelle und letztlich im Körper passiert irgendwie beteiligt.
Die mitochondrialen Dysfunktionen stellen somit eine enorme Herausforderung für Ärzte, Wissenschaftler und letztlich die Betroffenen dar.
Gegenwärtige Schätzungen kommen zu dem Schluss, dass etwa eine von 2000 Personen an einer mitochondrialen Dysfunktion leidet [1]. Diese Schätzungen berufen sich allerdings vorrangig auf Personen, welche auch eine Mutation in einem von 1300 Genen, welche im Zellkern oder einem von 37 Genen, welche in den Mitochondrien selbst kodiert sind, zeigen [1, 2]. Nicht berücksichtigt werden jedoch oft die Probleme im zellulären Gefüge, die ebenfalls die mitochondriale Leistungsfähigkeit beeinträchtigen [6]. Zu diesen gehören beispielsweise Probleme in den zuführenden Pathways, in Recycling-Mechanismen oder die Exposition zu toxischen Substanzen (wie Antibiotika oder Schwermetalle), welche die mitochondriale Leistungsfähigkeit beeinträchtigen können [7].
Der einschlägigen Literatur nach zu urteilen ist es gegenwärtig überhaupt sehr schwierig angemessene Therapien vorzuschlagen, denn z.B. einen Gendefekt ist momentan noch nicht korrigierbar [1, 8]. Solche Interventionen wurden zwar schon oft erprobt, jedoch ist eine flächendeckende sichere Anwendung noch in weiter Ferne [8]. Darüberhinaus wird es umso schwerer solche Probleme zu beheben je mehr Gene betroffen sind.
Vielleicht sollten wir uns aber erst einmal einen Überblick über mögliche Szenarien verschaffen, um zu verstehen woher die Probleme stammen und welche Möglichkeiten es geben könnte diese vielleicht zu vermeiden oder im Ernstfall mild zu behandeln.
Der gegenwärtigen wissenschaftlichen Literatur nach können primär fünf Hauptursachen für mitochondriale Dysfunktionen definiert werden:
- Mutationen der mitochondrialen DNA, welche mit der Herkunft verbunden sind und schon viele Jahrhunderte/Jahrtausende in einer bestimmten Volksgruppe vorhanden sind.
- Mutationen der mitochondrialen DNA, welche erst „kürzlich“ eingebracht wurden.
- Mutationen in der mitochondrialen DNA, welche man im Laufe des Lebens anhäuft.
- Mutationen von Genen in der Kern-DNA, die für mitochondriale Genprodukte kodieren. Und schließlich:
- Mutationen, die den Crosstalk zwischen Mitochondrien und Zellkern beeinflussen. Ich würde jedoch noch einen Schritt weitergehen und Punkt 6. definieren als:
- Störungen des Crosstalks zwischen Mitochondrien und anderen Zellorganellen, wie dem Endoplasmatischem Retikulum (ER). Außerdem ist meiner Meinung nach noch Punkt 7. wichtig, nämlich:
- Störungen im Metabolismus und in Signalling Pathways, die eine optimale Mitochondrienfunktion torpedieren.
Ich würde vorschlagen alle Probleme einzeln zu beschreiben und zu überprüfen inwiefern diese vermieden, gelindert oder ganz geheilt werden können.
Beginnen wir mit Punkt 1.: Mutationen, welche charakteristisch für bestimmte Volksgruppen sind.
Diese Mutationen haben sich innerhalb von Generationen im mitochondrialen Genom von Menschen eingenistet und konnten sich deshalb dort halten, weil sie u.a. einen Vorteil, keinesfalls aber einen dramatischen Nachteil zum Überleben darstellten [2]. Diese Ansammlungen von genetischen Besonderheiten in geographisch klar definierten Bevölkerungsgruppen, die sich von einem gemeinsamen Vorfahren ableiten, werden als Haplotypen bezeichnet [2, 4, 9].
 Abb.1 Haplotypen dieser Erde. Beachtet, dass es haufenweise verschiedene Haplotypen gibt. Das Bild soll den Text lediglich etwas auflockern. Wenn ihr mehr über eure potentielle „Haplotypenzugehörigkeit“ erfahren wollt, dann schaut einfach mal hier vorbei. Made by Chapper - unrestricted use allowed
Abb.1 Haplotypen dieser Erde. Beachtet, dass es haufenweise verschiedene Haplotypen gibt. Das Bild soll den Text lediglich etwas auflockern. Wenn ihr mehr über eure potentielle „Haplotypenzugehörigkeit“ erfahren wollt, dann schaut einfach mal hier vorbei. Made by Chapper - unrestricted use allowed
Sie waren oft Adaptationen an das vorherrschende Klima, die verfügbaren Nahrungsquellen und vieles mehr [2]. Einst beeinflussten die Umweltbedingungen sehr stark das Leben der Menschen. In Zeiten von Überfluss, guter Infrastruktur und hervorragender hygienischer wie medizinischer Versorgung, ist dies allerdings kein Thema mehr. Eher ist das Gegenteil der Fall und so kann sich beispielsweise kalorienreiche Nahrung für manche Menschen als sehr nachteilig herausstellen [4]. Doch auch Langlebigkeit wird unter anderem mit Haplotypen in Verbindung gebracht [2].
Wer mehr über Haplotypen erfahren möchte, der kann auf der Ancient mtDNA database vorbeischauen und sich vielleicht selbst sequenzieren lassen.
Werde ich bestimmt auch bald mal tun!
Behandelbar sind solche Mutationen ohne genetische Interventionen wahrscheinlich nicht. Der Grund ist einfach, dass diese Mitochondrien einer Selektion unterlagen und sich dabei gegen andere Mitochondrien durchsetzen konnten. Die Mitochondrien, welche ihr von eurer Mutter erhalten habt (Mitochondrien werden immer mütterlicherseits vererbt), sind demnach bzgl. diese Haplotypen reinerbig. Obwohl es mittlerweile Zweifel darin gibt [10].
Gleiches gilt prinzipiell auch für Punkt 2.: Mutationen, welche sich in vergleichsweise kurzen Zeiträumen ergeben haben.
Auch hier sind (der Literatur nach) genetische Eingriffe notwendig [1]. Die Kategorie umfasst, meiner Meinung nach, die schwerwiegendsten Probleme. Zu den „milderen“ Probleme dieser Art gehören z.B. Diabetes Mellitus Typ II, geringe Belastbarkeit bei sportlichen Anstrengungen oder Taubheit [2]. Alle anderen wirken sich in extremer Art und Weise auf das Leben der betroffenen aus. Beispiele hierfür sind u.a. MELAS. MELAS ist eine Krankheit welche sich u.a. durch Mitochondrien-assoziierte Hirnschäden (Enzephalopathien), starke Ansäuerung des Blutes (Laktische Azidose), sowie durch schlaganfallartige Episoden (Stroke) äußert. Verantwortlich ist hier, allem Anschein nach, oft nur eine einzige Mutation in einem einzigen mitochondrialen Gen für eine tRNA, welche die Aminosäure Leucin zu den mitochondrialen Ribosomen transportiert (Abb.2) [1, 2].
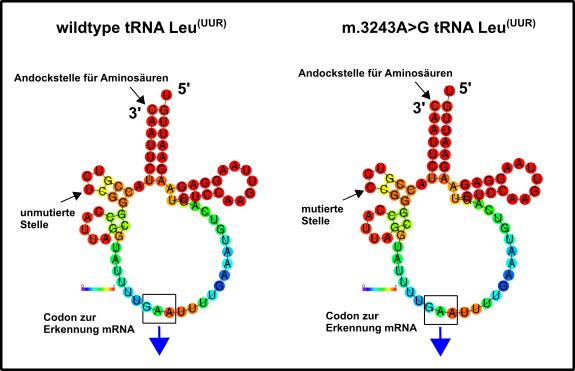 Abb.2 Mitochondriale tRNA für die Aminosäure Leucin. Links die Wildtyp-Variante, rechts die Variante, welche bei der Erkrankung MELAS mutiert sein kann. Die ursprüngliche mtDNA-Sequenz, welche diese tRNA kodiert fand ich hier. Danach habe ich zunächst die komplementäre Sequenz erstellt, weil die Transkription immer antiparallele Produkte hervorbringt. Dies gelang mir mit diesem Tool hier. Schließlich wurde das Produkt in RNA umgeschrieben, denn RNA trägt beispielsweise statt einem Thymin (T) ein Uracil (U). Dies umzuschreiben ist aber dank diesem Werkzeug hier kein Problem. Letztlich wurde mit dem RNAFold WebServer ein mögliches Modell der beiden tRNAs erstellt. Dargestellt ist die Tendenz zur Knüpfung von Bindungen. Aber auch die Entropieverteilung zeigte mir keinerlei gravierende Unterschiede an. Entweder ich muss dies künftig noch etwas ausgefeilter machen oder der Nachteil dieses Basenausstauschs ergibt sich durch direkte Wechselwirkungen, z.B. mit dem Ribosom, welche nach Mutation nicht mehr oder nur noch eingeschränkt gewährleistet sind. Made by Chapper - unrestricted use allowed
Abb.2 Mitochondriale tRNA für die Aminosäure Leucin. Links die Wildtyp-Variante, rechts die Variante, welche bei der Erkrankung MELAS mutiert sein kann. Die ursprüngliche mtDNA-Sequenz, welche diese tRNA kodiert fand ich hier. Danach habe ich zunächst die komplementäre Sequenz erstellt, weil die Transkription immer antiparallele Produkte hervorbringt. Dies gelang mir mit diesem Tool hier. Schließlich wurde das Produkt in RNA umgeschrieben, denn RNA trägt beispielsweise statt einem Thymin (T) ein Uracil (U). Dies umzuschreiben ist aber dank diesem Werkzeug hier kein Problem. Letztlich wurde mit dem RNAFold WebServer ein mögliches Modell der beiden tRNAs erstellt. Dargestellt ist die Tendenz zur Knüpfung von Bindungen. Aber auch die Entropieverteilung zeigte mir keinerlei gravierende Unterschiede an. Entweder ich muss dies künftig noch etwas ausgefeilter machen oder der Nachteil dieses Basenausstauschs ergibt sich durch direkte Wechselwirkungen, z.B. mit dem Ribosom, welche nach Mutation nicht mehr oder nur noch eingeschränkt gewährleistet sind. Made by Chapper - unrestricted use allowed
Jedem ist hoffentlich klar, dass Leucin eine wichtige Aminosäure ist, deren Abwesenheit einen korrekten Zusammenbau der mitochondrialen Proteine gar nicht oder nur unzureichend ermöglicht [11, 12]. Diese kleine Mutation hat demnach erhebliche Auswirkungen. Wenn ihr mich fragt sollten sich solche Erkrankungen aber in naher Zukunft sehr gut behandeln lassen. Ähnliches ist auch im Falle der Erkrankung MEERF gegeben, die noch viel zahlreichere Symptome aufweist, ebenfalls aber häufig auf eine einzelne Mutation in einer mitochondrialen tRNA reduziert werden kann [1]. Oberflächlich betrachtet scheint es, als ob Mutationen in mitochondrialen tRNAs den Löwenanteil der mitochondrialen Dysfunktionen in Kategorie 2 ausmachen. Die Menschen mit diesen Problemen sind häufig schon als Kinder betroffen, die Probleme verschlimmern sich, wie beim Charcot–Marie–Tooth-Syndrom, aber bis ins Erwachsenenalter immer mehr und mehr [1]. Wer Leute kennt, die an sowas leiden weiß, dass es oft ein mühseliger Kampf um etwas Lebensqualität ist. Nicht zu fassen, dass eine einzige falsche DNA-Base dafür verantwortlich sein soll. Einer meiner ehemaligen Diplomanden leidet auch an sowas, aber er ist ein Kämpfer. Respekt! Und Respekt natürlich auch gegenüber all den fleißigen Genetikern, die hoffentlich bald eine Möglichkeit gefunden haben, um dies zu korrigieren.
Dennoch frage ich mich ob die Annahme, dass erst genetische Eingriffe eine Linderung bringen könnten korrekt ist, denn immerhin verschlimmern sich die Probleme zum Erwachsenenalter hin. Die Mitochondrienerkrankung LHON beispielsweise führt erst im zweiten oder dritten Lebensabschnitt zu allmählicher Erblindung [3]. Irgendwas muss demnach im Zeitraum bis dahin im Körper passieren, was später diese Auswirkungen zeigt. Wir kommen später nochmal darauf zurück.
Kommen wir erstmal zu Kategorie 3.: Mitokrankheiten, die sich im Laufe des Lebens ansammeln. Sogenannte somatische Mutationen.
So hier wird es jetzt ziemlich spannend wie ich finde und hier ist es auch höchste Zeit mein Inkscape wieder richtig startklar zu machen. Prinzipiell diskutieren wir diese Problematik schon die ganze Zeit während ich diesen Blog hier mache und jedem Stammleser sollte klar sein was jetzt kommt. Richtig: Euer Life-Style. Wenn die Leute aus den ersten beiden Kategorien nun wirklich nix für ihr Leid können, so kommen wir jetzt in den Bereich des (potentiell) selbstverantworteten Leids. Lasst es mich kurz illustrieren.
(Ich beziehe mich in dieser Kategorie primär auf meine bereits angefertigten Post über Autophagie, sowie meine Reihe über gesundes Leben, eine Zusammenstellung findet ihr hier).
Dies sind eure Mitochondrien, wenn ihr jung, frisch und knackig seit:
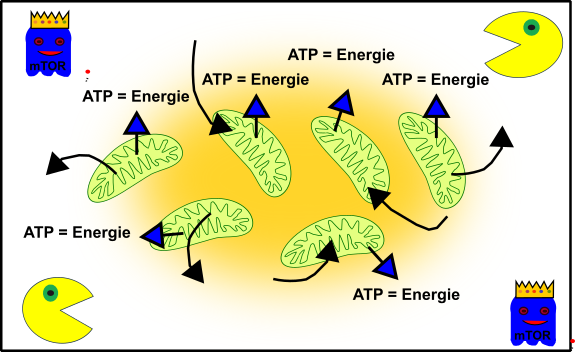 Abbildung 3: Vereinfachte Darstellung eures zellulären Gefüges in jungen Jahren. Eure Mitochondrien (grün), Autophagie (Packman) und fördernde Pathways (Gespenst in blau mit Krone). Made by Chapper - unrestricted use allowed
Abbildung 3: Vereinfachte Darstellung eures zellulären Gefüges in jungen Jahren. Eure Mitochondrien (grün), Autophagie (Packman) und fördernde Pathways (Gespenst in blau mit Krone). Made by Chapper - unrestricted use allowed
Eure Mitos produzieren eifrig Energie damit ihr die ganze Zeit unermüdlich rumrennen könnt‘. mTOR (das blaue Gespenst) lässt euren Körper schön wachsen und gedeihen. Und euer zellulärer Packman (Autophagie) passt auf, dass alles schön sauber und ordentlich ist. Die perfekte Symbiose, wenn ihr wollt. (Kleine Anmerkung an meine Neuleser, falls euch das gerade etwas bekloppt vorkommt, dann checkt bitte meine anderen Posts zum Thema, Danke!).
Nun aber werdet ihr älter und alles gerät etwas in Schieflage. Zum Beispiel habt ihr vielleicht etwas mehr Kummer, esst die ganze Zeit bei McDonalds, fangt an zu Rauchen und zu saufen oder Schlimmeres. Nun ist das Gefüge nicht mehr ganz so prickeln wie einst:
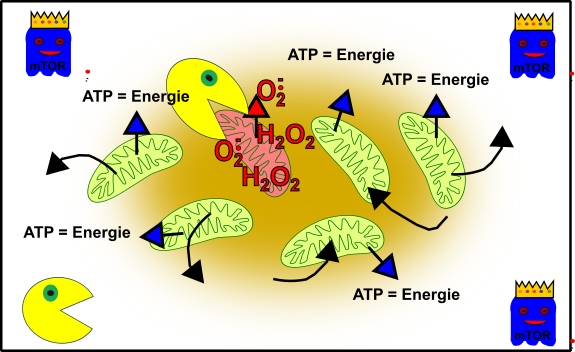 Abbildung 4: Vereinfachte Darstellung eures zellulären Gefüges im Verlauf eurer Adoleszenz. Eure Mitochondrien (grün), durch oxidativen Stress beschädigte Mitochondrien (rötlich), Autophagie (Packman) und fördernde Pathways (Gespenst in blau mit Krone). Made by Chapper - unrestricted use allowed
Abbildung 4: Vereinfachte Darstellung eures zellulären Gefüges im Verlauf eurer Adoleszenz. Eure Mitochondrien (grün), durch oxidativen Stress beschädigte Mitochondrien (rötlich), Autophagie (Packman) und fördernde Pathways (Gespenst in blau mit Krone). Made by Chapper - unrestricted use allowed
Der erhöhte Stress in euren Zellen hat in einigen Mitochondrien zu erheblichen Schäden geführt. Oxidativer Stress (z.B. H2O2) hat diese Mitochondrien außer Gefecht gesetzt. Aber keine Panik! Euer zellulärer Packman (Autophagie) bereinigt die Sache und weiter geht’s. Jeder weiß, dass es stets immer schwieriger wird im Leben und auch eure Zellen haben mit den Jahren mehr und mehr zu kämpfen. Nicht nur, dass euer zellulärer Packman zunehmend fauler wird [13], nein auch mTOR zeigt mehr und mehr sein wahres Gesicht und verhindert nicht nur, dass Zellen, die am Ende sind endlich sterben [14], im Gegenteil, mTOR arbeitet aktiv gegen unseren lieben Packman [15]:
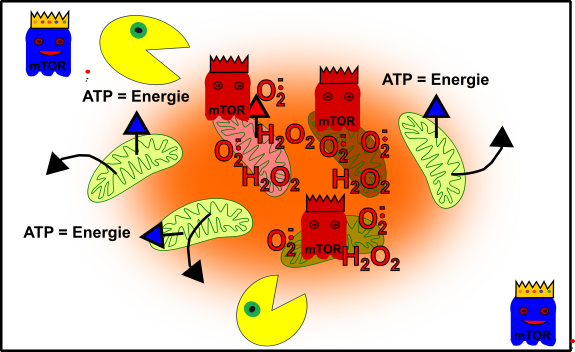 Abbildung 5: Vereinfachte Darstellung eures zellulären Gefüges im Erwachsenenalter. Eure Mitochondrien (grün), durch oxidativen Stress beschädigte Mitochondrien (rötlich), Autophagie (Packman), fördernde Pathways (Gespenst in blau mit Krone) und aus dem Gelichgewicht geratene fördernde Pathways (rote Gespenster mit Krone). Made by Chapper - unrestricted use allowed
Abbildung 5: Vereinfachte Darstellung eures zellulären Gefüges im Erwachsenenalter. Eure Mitochondrien (grün), durch oxidativen Stress beschädigte Mitochondrien (rötlich), Autophagie (Packman), fördernde Pathways (Gespenst in blau mit Krone) und aus dem Gelichgewicht geratene fördernde Pathways (rote Gespenster mit Krone). Made by Chapper - unrestricted use allowed
Nun beginnen die Probleme. Du bist schwach, ausgebrannt, überall Schwierigkeiten. Keine Leistung, keine Kraft, ständig hast du irgendwas. Du denkst: „Diese oder jene Pille wird schon helfen!“ „Ein kleiner Absacker hat noch niemandem geschadet!“ „Was kann an ein paar aufmunternden Süßigkeiten schon verkehrt sein, wo ich doch heute so niedergeschlagen bin?“ In Wahrheit verschlimmern all diese Interventionen aber alles noch viel mehr, denn mTOR hat nur darauf gewartet, um euren zellulären Packman noch mehr zu unterdrücken. Der Terror nimmt seinen Lauf. Die Mitochondrien, die einen totalen Knacks haben, werden nun nicht einfach beseitig, vielmehr vermehren sie sich auch noch [4]. Und so habt ihr am Ende die Mutationen, welche schwerkranke Menschen in ihren Mitochondrien von Anfang an tragen (+X), ebenfalls in euerem Bestand.
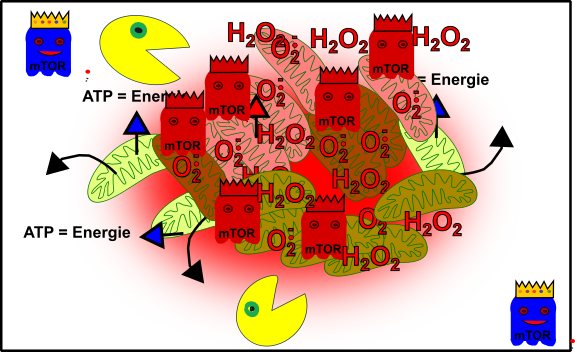 Abbildung 6: Vereinfachte Darstellung: Euer zelluläres Gefüge ist im Eimer! Eure Mitochondrien (grün), durch oxidativen Stress beschädigte Mitochondrien (rötlich), Autophagie (Packman), fördernde Pathways (Gespenst in blau mit Krone) und aus dem Gleichgewicht geratene fördernde Pathways (rote Gespenster mit Krone). Made by Chapper - unrestricted use allowed
Abbildung 6: Vereinfachte Darstellung: Euer zelluläres Gefüge ist im Eimer! Eure Mitochondrien (grün), durch oxidativen Stress beschädigte Mitochondrien (rötlich), Autophagie (Packman), fördernde Pathways (Gespenst in blau mit Krone) und aus dem Gleichgewicht geratene fördernde Pathways (rote Gespenster mit Krone). Made by Chapper - unrestricted use allowed
Herzlichen Glückwunsch! Du hast erfolgreich deine Gesundheit ruiniert!
Und nun? Wie geht’s weiter? Tja, ich kann nicht alles eine Million Mal wiederkäuen, check meine anderen Posts zu vergleichbaren Themen und du wirst eine Antwort finden.
Punkt 4. Ist wieder weniger selbstverschuldet: Mutationen in der Kern-DNA.
Dies ist ein besonders spannender Aspekt wie ich finde, denn wie ihr bereits aus dem ersten Teil dieser Serie wisst, haben die Mitochondrien nach ihrer „Übernahme“ (Endosymbiose) zahlreiche Gene in den Kern ausgelagert [1, 2, 4, 16]. Sage und Schreibe 1300 Stück(!). Nur 37 Gene sind in den Mitochondrien selbst verblieben (wenn auch die Wichtigsten). Nichtdestotrotz sind diese 1300 Gene nicht minder notwendig [2]. Die Komplexe der Atmungskette werden beispielsweise sowohl von Genen kodiert, die direkt in den Mitochondrien als auch im Kern liegen. Aber es gibt auch Ausnahmen: Kompex II der Atmungskette wird beispielsweise ausschließlich im Kern kodiert [2]. Kein Wunder also, dass es hier ebenfalls Problemen kommen kann.
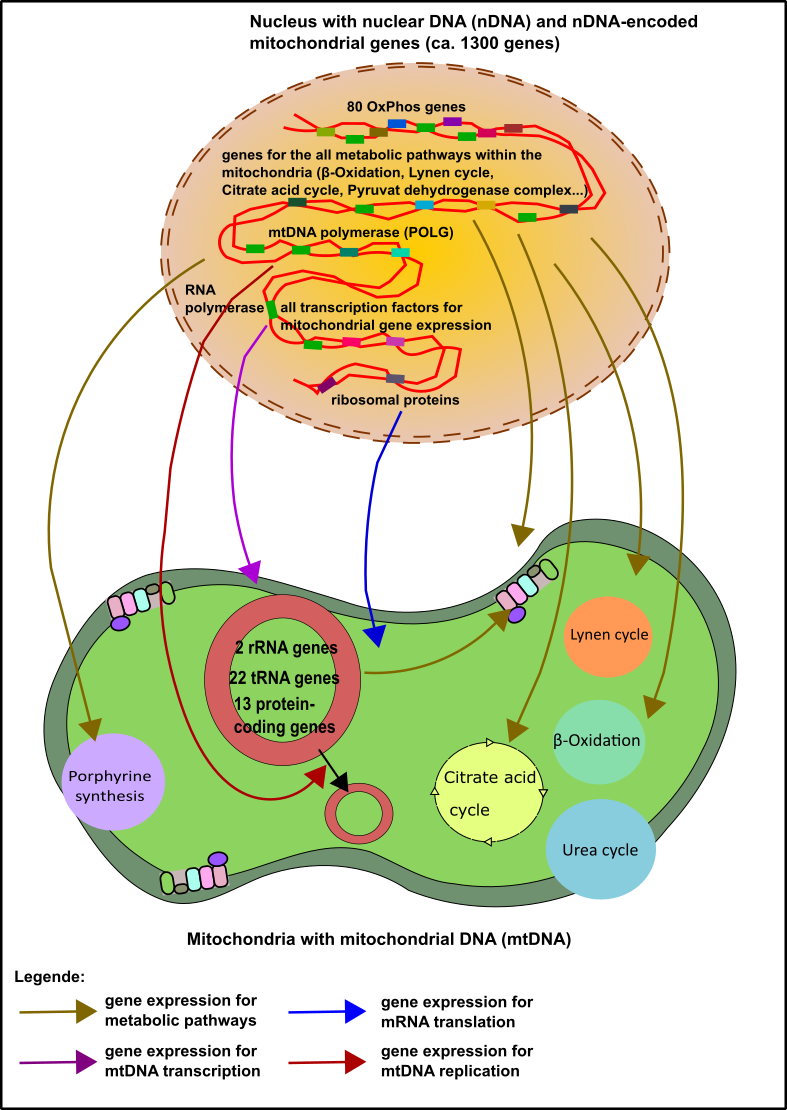 Abbildung 7: Die Mehrheit der mitochondrialen Gene sind im Kern kodiert. Diese „Kerngene“ sind für alles notwendig, sowohl für sämtliche Enzymatik, als auch für die DNA-Replikation des mitochondrialen Genoms (mtDNA). Darüber hinaus arrangieren die im Kern gespeicherten Gene die Transkription der mtDNA-Gene und die Translation der mRNAs, welche die mtDNA hervorbringt. Made by Chapper - unrestricted use allowed
Abbildung 7: Die Mehrheit der mitochondrialen Gene sind im Kern kodiert. Diese „Kerngene“ sind für alles notwendig, sowohl für sämtliche Enzymatik, als auch für die DNA-Replikation des mitochondrialen Genoms (mtDNA). Darüber hinaus arrangieren die im Kern gespeicherten Gene die Transkription der mtDNA-Gene und die Translation der mRNAs, welche die mtDNA hervorbringt. Made by Chapper - unrestricted use allowed
Einen Vorteil hat der Kern allerdings: Er ist weniger stark anfällig für Mutationen als die DNA der Mitochondrien! Warum? Nun so richtig weiß das niemand. Sicherlich spielt es eine entscheidende Rolle, dass Mitochondrien die Nummer 1-Adresse für die Entstehung von Sauerstoffradikalen & Co. bzw. oxidativen Stress (Reaktive Oxygen Spezies, ROS) sind [17-19]. Da die Mito-DNA (mtDNA) direkt daneben liegt, ist es klar, dass diese umso schneller mutiert wird. Außerdem ist das Mitochondriengenom viel kleiner, d.h. wenn eine ROS vorbeikommt, dann trifft sie umso schneller auch ins Schwarze. Bedenkt bitte, dass das Mitochondrien-Genom fast durchgängig mit kodierenden Sequenzen voll ist, die Kern-DNA hingegen besteht zu über 98% aus jenem Material was nicht-kodierend ist [20]. Vieles davon ist landläufig als Junk, also Müll bekannt ist (zu Unrecht übrigens) [20, 21].
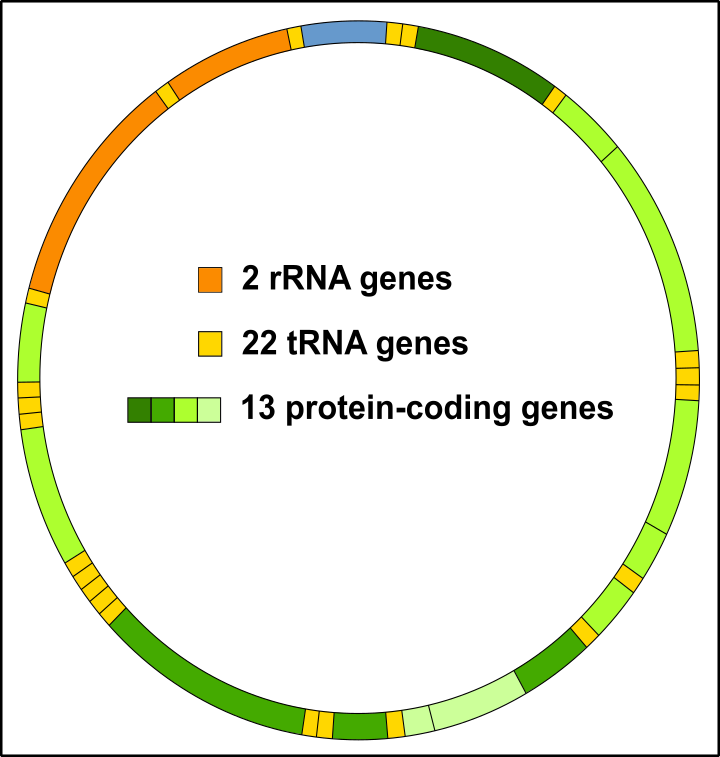 Abbildung 8: Das Mitochondriengenom (mtDNA) ist dichtgepackt mit Genen. Jeder Schuss ist somit ein Treffer! [Lizenzfrei CC BY-SA 3.0]
Abbildung 8: Das Mitochondriengenom (mtDNA) ist dichtgepackt mit Genen. Jeder Schuss ist somit ein Treffer! [Lizenzfrei CC BY-SA 3.0]